


微生物多样性测序介绍
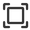
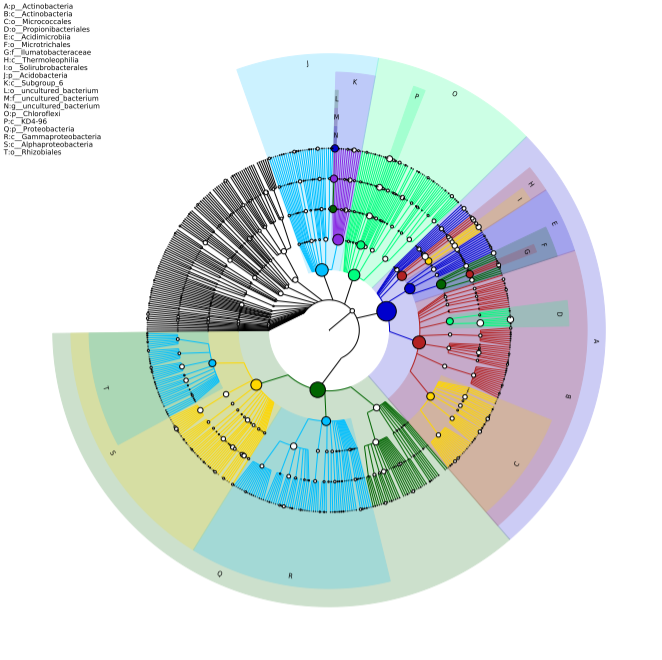
微生物多样性测序,又称扩增子测序,是利用二/三代测序平台,以细菌/古菌16S rRNA基因可变区、真菌18S rRNA基因可变区或真菌ITS(Internal transcribed spacer)内转录间隔区等微生物特征序列(Signature sequence)为靶点,通过检测序列的变异和数量,反映菌群中各类微生物的种类和丰度,从而快速解码菌群物种分布特征,阐明样本间多样性和组成差异。
技术服务路线



送样须知
1. 请务必认真填写各项内容
2. 寄送样品时请您附带一份送样单
3. 如样品质检不合格将收取部分提取费用,请悉知
4. 不接受具有致病性的样品。如果您的菌株带有致病性
5. 建议每样检测3个以上生物学重复(按多个样品收费)
需要提供
1. 污泥、土壤采样后置于无菌离心管内,短期可置于-20度,长期置于-80度保存,低温寄送,样品量3g+;
2. 动物粪便及肠道内容物,由于动物体内多种酶类可能会发生生物降解,取样器材尽量灭菌,用无菌离心管装样后寄送,长期保存于-80,干冰寄送,样品量不足时可以多次取样收集,样品量根据具体动物类型决定;
3. 水体样品可选用0.22 μm或者0.45 μm的滤膜过滤后寄送滤膜,一般一次送1-2张/样。
一般做二代测序(即扩增子测序)可以鉴定到属,三代一般是测菌的全长,信息量更多一些,有一些种属可以精确到种的信息,但是个别亲缘关系很近的,不一定能鉴别开。
真菌、细菌、古菌全长引物包含区段引物序列(V3V4,IT1区等),全长可以更准确的物种分类,更多的物种鉴定,能精确定位到“种”水平。
全长测序使用三代测序平台。基于PacBio 测序平台测序,相比较二代测序只选择 1-2 个高变区进行测序,三代可以获得全长的序列,更多变异区信息可以提高物种鉴定的分辨率,还可以提高对于群落组成的鉴定的精度,更加真实的还原样本中微生物群落结构。
①普通16S扩增子测序: 一般是通过对16S V3或V4区(大概200-300bp)PCR扩增建库后,进行高通量测序,选择这两个区域的原因是可检测的物种多样性更丰富,并且可重复性强,二代测序的读长可达到500bp,检测结果无需拼接。缺点是由于序列长度有限,对样品的鉴定一般只能精确到属。
②16S全长扩增子测序:16SrDNA的全长大约为1500bp,通常来说足够鉴定到种,其实现有两种方法:其一是通过二代测序,不同于V3V4区测序,16S全长超过了二代测序的有效读长,因此需要对结果进行拼接,此外,必须提高16S全长序列的质量,测序结果才能精确,因此对技术人员的操作要求也较高;其二是通过三代测序,它的优势是拥有超长的读长,16S全长测序结果无需拼接,其缺点就是错误率高,通量低,成本昂贵。
③宏基因组测序:是对样本中全部基因进行测序注释,能鉴定微生物到种水平甚至菌株水平,宏基因组测序在16S测序分析的基础上还可以解释环境样本中的功能组成及代谢通路等信息,主要用于深入挖掘环境样本功能层面的信息。它的技术原理是将微生物基因组DNA随机打断成较小片段,然后在片段两端加入通用引物进行PCR扩增测序,再通过组装的方式,将小片段拼接成较长的序列,由于宏基因组测序结果数据量巨大,耗时更长,操作流程也更复杂,因此收费也更高。
一个细菌分类地位的最终确定,需要做多相分类分析,包括形态,生理生化,进化和分子等特征,以后还会包括基因组分析。16S的数据只是一个参考值。







Copyright © 2023 - 2026 广州科奥信息技术股份有限公司 All Rights Reserved